Experimental and Clinical Pharmacology
Inside the isomers: the tale of chiral switches
- Andrew Somogyi, Felix Bochner, David Foster
- Aust Prescr 2004;27:109-13
- 1 March 2004
- DOI: 10.18773/austprescr.2004.039
Chiral drugs are made up of molecules with the same chemical structure, but different three-dimensional arrangements. Modern manufacturing has enabled the development of products containing a single molecular arrangement. The development of these single enantiomers from chiral drugs is known as chiral switching. Enantiomers of the same drug can have different pharmacodynamic and pharmacokinetic properties. This may translate into potential health benefits, such as an improved safety margin, if one of the enantiomers has more favourable therapeutic and pharmacokinetic characteristics. However, some chiral switching has resulted in unpredicted toxicity and the withdrawal of the enantiomer from the market or a halt in its development. Drug companies are increasingly using chiral switching as a marketing strategy, but before prescribers switch to single enantiomer drugs they should look for evidence from well-conducted clinical trials that shows the chiral switch is cost-effective and improves the outcomes for patients rather than patents.
In the Introduction, salmeterol was cited as a single enantiomer drug, however, it is currently marketed as the racemate - noting that the R enantiomer is the active species.
Have you ever tried putting your left shoe on your right foot or your right glove on your left hand? Unless you intend to destroy the function of this apparel, you know you will not succeed. Your left and right hands and feet are non-superimposable mirror images of one another. Chemicals including drugs can behave in a similar way. Many drugs consist of a mixture of left-and right-handed molecules (enantiomers), but there is an increasing trend for the pharmaceutical industry to develop and market products containing only the left-or right-handed molecule.1 While many of these single enantiomer drugs (such as sertraline and salmeterol) are new chemical entities, others have been developed from currently marketed drugs which are a mixture of different enantiomers (racemates). For example, esomeprazole is an enantiomer of the racemate omeprazole. The term chiral switching has been coined to describe the development of single enantiomers from old racemate drugs.
| Enantiomer: | one of a pair of stereoisomers that are non-superimposable mirror images of one another and therefore have a different 3-dimensional configuration |
| Isomers: | compounds with the same molecular formula but with different 3-dimensional configuration |
| Racemate: | a mixture of two enantiomers, usually in one-to-one ratio |
If a drug has a centre of asymmetry (usually a carbon atom with four different substituents), then it can exist as two non-superimposable left-handed and right-handed mirror images, also known as enantiomers. A racemate is a mixture of equal amounts of these two enantiomers. Many drugs are marketed as racemates. They are said to be chiral drugs (from the Greek word for hand, cheir).
There are different methods for naming enantiomers. The definitive way is to use the prefix (R)-(right hand) and (S)-(left hand). Other prefixes are (+) and (–) or D and L. An example is ibuprofen (Fig. 1) which, as marketed in Australia, contains an equal amount of (R)-ibuprofen and (S)-ibuprofen.
Enantiomers have identical physical and chemical properties such as molecular weight, solubility and melting point. The only difference is their three-dimensional spatial configuration.
Most drugs obtained from nature are chiral, but in nature only the biologically active enantiomer is synthesised. For example, the poppy plant Papaver somniferum only synthesises the pain relieving (–)-(5R,6S,9R,13S,14R)-morphine. As morphine has a demanding chemical structure with five asymmetric centres, the technical difficulties and costs associated with chemically manufacturing large amounts for therapeutic use are such that it is more economically viable for companies to extract the morphine for the world market from poppies, rather than to artificially synthesise it. However, for many other chiral drugs, synthesis of the individual enantiomers is now economically feasible.
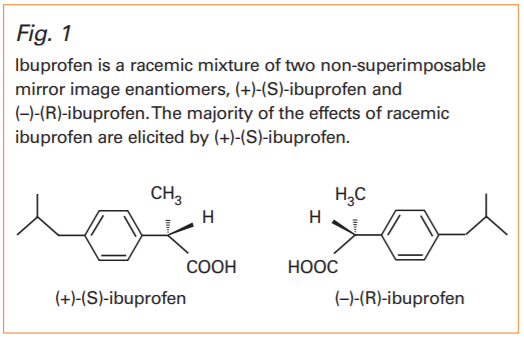
Ibuprofen is a racemic mixture of two non-superimposable mirror image enantiomers, (+)-(S)-ibuprofen and (–)-(R)-ibuprofen. The majority of the effects of racemic ibuprofen are elicited by (+)-(S)-ibuprofen.
The interactions in the body between a drug and the proteins which elicit therapeutic or adverse effects and eliminate the drug require a specific three-dimensional configuration of drug and protein.
Since enantiomers have different three-dimensional configurations, the pharmacodynamics and pharmacokinetics of the two enantiomers which make up a racemic drug can be quite different. The differences often depend on whether the centre of asymmetry of the drug is in close proximity to the points of attachment to the protein. For example:
The so-called inactive enantiomer (one that has much less affinity for the drug's target site) is not necessarily an inert substance with no effects in vivo. For example, the cardiotoxicity of bupivacaine is mainly associated with the (R)-enantiomer, the psychomimetic effects of ketamine are more associated with the (R)-enantiomer, and (S)-baclofen antagonises the effects of (R)-baclofen. The beneficial effects of a drug can therefore reside in one enantiomer, with its paired enantiomer having:
As the distribution and elimination of drugs from the body also involves their interaction with proteins, then the pharmacokinetics of enantiomers can also be different. For example:
These differences in clearance and volume of distribution translate into differences in half-life. For example the half-life of (S)-fluoxetine is one quarter that of (R)-fluoxetine. In addition, these pharmacokinetic properties can be modified in a stereoselective manner by disease, genetics, ethnicity, age and other drugs. Finally, the enantiomers of some drugs such as warfarin can be metabolised by different enzymes.
There are several possible health benefits to chiral switching. They include:
As some racemic drugs were patented without separate patents for each enantiomer, some companies have seized the opportunity to develop and market or license single enantiomers of marketed chiral drugs (for example an American company now markets (R)-salbutamol). Another commercially driven reason for chiral switches is the impending expiry of the patents of some 'blockbuster' racemic drugs. The manufacturers have developed and marketed the single enantiomer with a view to extending the patent franchise and protecting themselves from competitors who produce generic copies of the racemate.2
Obtaining marketing approval for a chiral switch usually requires relatively few new studies to be conducted if the racemate is already marketed. The single enantiomer can be ready for launch before the patent for the racemate expires and before the marketing of any generics (which tend to substantially drive down the cost of the racemate).
Many single enantiomer chiral switches have recently received marketing approval in Australia or are likely to be submitted for approval. These include single enantiomers of omeprazole, bupivacaine, citalopram, ofloxacin, salbutamol, ketamine, methylphenidate, cetirizine and oxybutinin. In most cases, the manufacturer has claimed specific advantages over the racemate, particularly decreased incidences and severity of adverse effects. These claims need to be confirmed in clinical trials with sufficient power to show any clinically significant advantages.
In some cases, chiral switching has been of no benefit. For example, the clinical development of (R)-fluoxetine for depression (based on a more acceptable half-life and less propensity for significant drug-drug interactions) was stopped because of a small but statistically significant prolongation of the QT interval with high doses. Dilevalol was thought to have advantages over labetalol, but was removed from the Japanese market because of hepatotoxicity.
A recent addition to the Pharmaceutical Benefits Scheme is esomeprazole, the (S)-enantiomer of omeprazole. All proton pump inhibitors exist as two inactive enantiomers (prodrugs) that are converted to active moieties which equally inactivate the H+ /K+ -ATPase pump. Both enantiomers of omeprazole are equipotent, however, their metabolism is different. (R)-omeprazole is mainly metabolised by the polymorphic CYP2C19 enzyme. There is a 7.5-fold difference in the systemic exposure to (R)-omeprazole in patients who are poor metabolisers compared to extensive metabolisers. With (S)-omeprazole this difference is reduced to about three-fold so it was argued that use of esomeprazole would be associated with less inter-individual variability in efficacy. However, there are few data to support this theoretical advantage3 , especially when only 3% of the Caucasian population are poor metabolisers. There may be a benefit in the Asian population where the incidence of poor metabolisers is about 20%. A rationale for chiral switching to esomeprazole might therefore be based on ethnic differences in metabolism.
The selective serotonin reuptake inhibitor activity of citalopram and its active metabolites resides mainly in the (S)-enantiomer. This enantiomer and its metabolites are eliminated slightly faster from the body than the (R)-enantiomer and its metabolites. In overdose, there is a concern about the potential for sudden death, possibly related to QT prolongation due to a secondary metabolite formed from (R)-citalopram. (S)-citalopram (escitalopram) was therefore developed with the aim of a better harm: benefit ratio compared to (R)-citalopram. However, this potential clinical advantage remains to be clinically proven.
Drug development is becoming longer and more complex, while marketing is increasingly competitive. Differences between single enantiomers and racemates are likely to become the focus for aggressive promotion of the 'new' entity. Regulatory authorities and independent sources of drug information (Australian Medicines Handbook, Australian Prescriber, Therapeutic Guidelines, National Prescribing Service) need to be provided with good evidence, from well-conducted clinical trials and appropriate pharmacoeconomic studies, that chiral switches have advantages for the prescriber and the consumer. The future will see not only more chiral switches but metabolite switches and metabolite-chiral switches providing fertile ground for patent lawyers and clinical pharmacologists.
Ariens EJ. Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. Eur J Clin Pharmacol 1984;26:663-8.
Burke D, Henderson DJ. Chirality: a blueprint for the future. Brit J Anaesth 2002;88:563-76. Eichelbaum M,Testa B, Somogyi A, editors. Stereochemical aspects of drug action and disposition. Handbook of experimental pharmacology Vol 153. Berlin, NewYork: Springer; 2003.
Pang YT. Stereoisomerism in drug molecules. Aust Prescr 1989;12:19-22.
Williams KM, Day RO. Clinical applications of enantiomeric drugs. Aust Prescr 1989;12:22-5.
The authors have received funding for their research in this field from the National Health and Medical Research Council, and the US National Institutes of Health/National Institute on Drug Abuse.
Department of Clinical and Experimental Pharmacology, University of Adelaide, Adelaide
Department of Clinical and Experimental Pharmacology, University of Adelaide, Adelaide
Department of Clinical and Experimental Pharmacology, University of Adelaide, Adelaide
